Kinetics of formation/dissociation of biomolecular complexes¶
Recent results from our group show that for a system of \(\sim 10^3-10^5\) atoms all-atom MD simulations in implicit solvent run ~100-150-times faster on a GPU than on a single CPU core. This opens up new avenues of research, and makes it possible to study the kinetics of formation of ligand-receptor complexes, which occurs on the computationally accessible \(\sim 10-100 \mu s\) timescale. We take advantage of the computational speedup, obtained on a GPU, to explore the kinetics of formation of protein-protein (ligand-receptor) complexes. The two main systems, studied in our reseacrh group, are the so called A:a and B:b knob-hole complexes. Formation and rupture of the A:a and B:b knob-hole bonds are the strongest interactions in fibrin(ogen), which are the driving force for fibrin oligomerization. We collaborate with Dr. Weisel group to explore the molecular mechanisms underlying force-driven dissociation and formation of the A:a and B:b knob-hole complexes. We are hoping be able to predict the dependence of binding rates as a function of concentrations of interacting species (kinetic curves), and to develop new theory for formation of biomolecular complexes. Another important direction in our research group is to study the kinetics and mechanisms of interactions of the fibrinogen (ligand) and integrin \(\alpha II b \beta 3\) (receptor) molecules (see Figure). These interactions are important, e.g., in hemostasis and thrombosis (blood clotting). We are also exploring the kinetics and thermodynamics of hydrophobic interactions using simple models, such as short linear hydrocarbon chains (\(C_{10}-C_{10}\)). Hydrophobic interactions play a central role in many chemical, biological and technological processes. For example, the first step in protein folding is associated with the so called “hydrophobic collapse”, i.e., spontaneous formation of energetically favorable contacts among the hydrophobic residues.
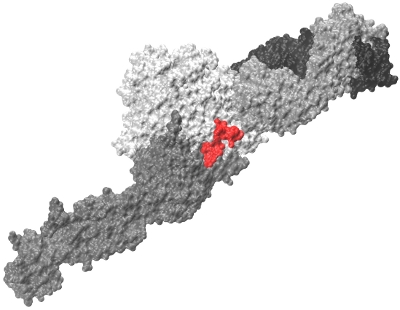
Figure: Fibrinogen \(\gamma C\) peptide (red) bound to integrin \(\alpha II b \beta 3\) headpiece receptor.
Kinetics of the A:a and B:b knob-hole interactions in fibrin¶
Formation of insoluble fibrin polymer from its soluble precursor protein, fibrinogen, is the final stage of blood clotting. The irreversible conversion of fibrinogen to fibrin occurs in three steps: (1) the limited proteolysis of fibrinogen by thrombin, which is followed by (2) the self-assembly of newly formed fibrin monomers into fibrin oligiomers, and by (3) the covalent crosslinking of assembled fibrin oligomers to strengthen the clot. The cleavage of the fibrinopeptides FpA and FpB occurs in the central \(N\)-terminal part of the fibrinogen molecule called the E-region. This results in the exposure of a pair of newly formed binding fragments ‘A’ and ‘B’. These binding determinants, also called ‘A’-knobs and ‘B’-knobs, interact with the complementary binding sites ‘a’ and ‘b’, respectively, which are called ‘a’-holes and ‘b’-holes, located in the \(\gamma\)-modules and \(\beta\)-modules, respectively, of the D-regions at the ends of the fibrinogen molecule (see Figure). We perform all-atom Molecular Dynamics (MD) simulations of the forced rupture of A:a and B:b knobe-hole bonds to explore the most important interactions for fibrin polymerization process. To speedup simulations we use traditional MD simulations in explicit solvent (water) on the CPU machines utilizing standard software packages, such as NAMD and GROMACS. In addition, we run MD simulations in implicit water fully implemented on graphics processors (GPUs), using the simulation methods recently developed in our lab. We plan to obtain full information about the kinetics and thermodynamics of the knob-hole interactions, in order to map out the free energy landscape underlying the A:a and B:b knob-hole interactions, i.e., to estimate the free energy barriers and “working distances” for binding and for unbinding. We also analyze how the mechanism of the A:a and B:b knob-hole interactions changes with the experimental conditions, such as temperature (T) and pH, and how single site mutations in the ‘a’-hole affect the strength of A:a interactions subject to mechanical factors such as a tensile force.
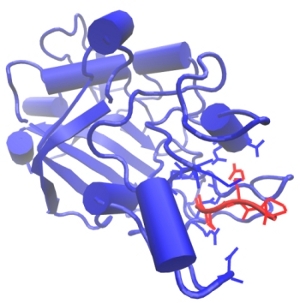
Figure: The biomolecular A:a knob-hole complex formed by binding of the knob ‘A’ (red) to the hole ‘a’ (blue). Also shown are the side chains of the most important binding determinants.
Kinetics of hydrophobic interactions¶
The hydrophobic effect is related to the disruption of the hydrogen bond network in water upon addition of a hydrophobic solute molecule as compared with the hydrogen bond network in pure water (see Figure). Interestingly, the details of the water structure around a hydrophobic solute depend on the size of the hydrophobic species. Although the nature of hydrophobic forces is well understood, and has been characterized, yet, little is known about the kinetics of hydrophobic interactions, i.e. the kinetics of formation and rupture of “hydrophobic bonds”, and how the kinects and the underlying mechanism change with the size (length) and geometry (linear molecule versus planar molecules) of hydrophobic species. Recent advances in Atomic Force Microscopy (AFM) have made it possible to probe directly the strength of hydrophobic interactions between single small hydrophobic molecules, such as hexadecane molecules (\(C_{16} H_{34}\)). This opens up a new direction of research, including the experimental and theoretical studies of the kinetics of hydrophobic interactions at a single molecule level. We employ all-atom Molecular Dynamics simulations in explicit water to investigate the mechanism for the forced rupture of “linear \(C_{16} H_{34}-C_{16} H_{34}\) bonds” and “planar \(C_{60}-C_{60}\) bonds”. We also explore the reverse process of the formation of \(C_{16} H_{34}-C_{16} H_{34}\) bonds and \(C_{60}-C_{60}\) bonds. The computational studies of formation and dissociation of the “hydrophobic bonds” is a formidable task, due to the fact that these are weak interactions, and, hence, many simulation runs should be performed to obtain any statistically significant information. We hope to estimate the free energy barriers and “working distances” for hydration, dewetting and complete dissociation of the hydrophobic bonds.

Figure: Hydrophobic interactions between two hexadecane molecules (\(C_{16} H_{34}-C_{16} H_{34}\)). Also shown are the water molecules surrounding the hydrophobic contact.